Expérience in silico
Comme pour d’autres types de simulations numériques, une simulation à l’échelle atomique peut s’apparenter au jumeau numérique d’une expérience menée en laboratoire. Elle cherche donc d’abord à reproduire de la façon la plus réaliste possible les conditions expérimentales telles que la température ou la pression, en adoptant un modèle à l’image du système étudié. Ainsi, un matériau solide ou un liquide n’est pas simplement décrit par un modèle incluant un nombre réduit d’atomes mais est soumis à des conditions aux limites périodiques lui conférant une taille infinie. Cette expérience dite in silico a pour objectif de produire des informations précieuses sur les mécanismes de processus physiques ou chimiques, leur cinétique, les propriétés structurales ou thermodynamiques du système, ou encore de fournir des données permettant d’interpréter des expériences de spectroscopie.
Approche classique
Au lieu de calculer explicitement la surface d’énergie potentielle électronique nécessaire à ce type de simulation, l’approche classique consiste à la représenter par un ensemble de termes simples, non quantiques, formant un champ de force. Evidemment, le gain de temps important obtenu par cette simplification est à mettre en balance avec l’exactitude de la prédiction du modèle, dont les paramètres sont obtenus de manière empirique (et donc dans des conditions thermodynamiques données pas automatiquement transposables à d’autres situations). Ainsi, les termes décrivant des atomes liés sont typiquement des oscillateurs harmoniques semblables à celui d’une masse suspendue à un ressort, et les interactions entre atomes non liés sont décrites par la somme des potentiels électrostatique (défini par des charges atomiques empiriques) et de Lennard-Jones, qui rend compte à la fois de la répulsion électronique à courte distance et de l’attraction de Van de Waals à longue distance,
![E_{LJ}=\sum_{ij}4\epsilon_{ij}\left[\left(\frac{\sigma_{ij}}{r_{ij}}\right)^{12}-\left(\frac{\sigma_{ij}}{r_{ij}}\right)^{6}\right]](https://s0.wp.com/latex.php?latex=E_%7BLJ%7D%3D%5Csum_%7Bij%7D4%5Cepsilon_%7Bij%7D%5Cleft%5B%5Cleft%28%5Cfrac%7B%5Csigma_%7Bij%7D%7D%7Br_%7Bij%7D%7D%5Cright%29%5E%7B12%7D-%5Cleft%28%5Cfrac%7B%5Csigma_%7Bij%7D%7D%7Br_%7Bij%7D%7D%5Cright%29%5E%7B6%7D%5Cright%5D&bg=ffffff&fg=000&s=0&c=20201002)
Dynamique moléculaire ab initio
Si l’on souhaite conserver une description quantique des degrés de liberté électroniques, et ainsi être en capacité de décrire des ruptures et formation de liaisons chimiques, l’approximation de Born-Oppenheimer doit être prise à bras-le-corps. Les noyaux sont alors propagés au cours de la simulation, et l’énergie électronique est calculée à chaque pas de temps en résolvant l’équation de Schrödinger correspondante de manière approximative. En pratique, la théorie de la densité fournit un bon compromis entre rapidité et fiabilité du calcul.
A cette méthode directe, on préférait il y a quelques années l’approche imaginée par Car et Parrinello, qui consistait à propager dans le temps simultanément les degrés de liberté nucléaires et électroniques à partir de conditions initiales. Celle-ci permettait une conservation de l’énergie totale du système qui ne pouvait être maintenue de façon satisfaisante dans l’approche directe de Born-Oppenheimer. Aujourd’hui, des algorithmes utilisent l’historique des orbitales de Kohn-Sham déjà calculées pour extrapoler les suivantes (de manière réversible), ce qui a entièrement corrigé ce problème. L’essor des simulations ab initio peut néanmoins être observé en traçant le nombre de citations de l’article de Car et Parrinello au cours des années.
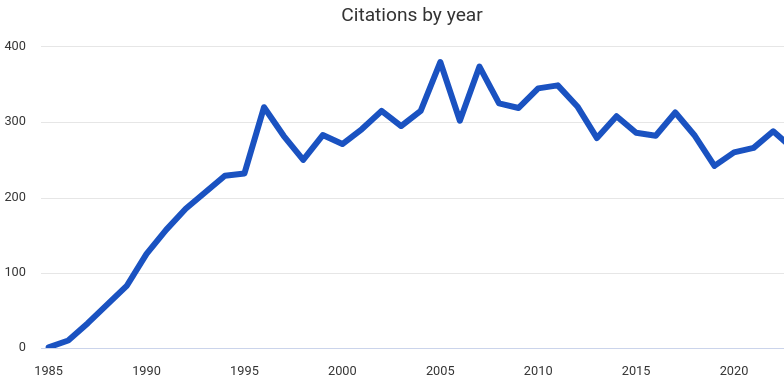
Si l’approche classique utilisant des champs de force demeure nettement plus rapide et permet l’étude de systèmes de très grande taille, les simulations ab initio de systèmes de plusieurs centaines d’atomes sur des durées approchant la nanoseconde sont désormais possibles. Ce progrès a notamment été rendu possible grâce à l’augmentation des performances des supercalculateurs.


Laisser un commentaire